
序にかえて
細胞老化研究の転換点:慎重な研究のすゝめ
原 英二
(大阪大学微生物病研究所分子生物学分野)
はじめに
senescence-associated secretory phenotype(SASP:細胞老化随伴分泌現象)の再発見によって,細胞老化の概念にパラダイム・シフトが生じた1).それまで不可逆的な細胞分裂の停止が細胞老化の主な役割とされていたが,老化細胞がさまざまな分泌因子を介して周囲の細胞に影響を与えることが明らかになり,細胞老化の概念が大きく変化した2).また,同時に,細胞老化の作用を正確に把握することが難しくなったことも事実である.こうしたなか,相反する実験結果や再現性が確認できない実験結果が学術論文として相次いで報告され,現在,細胞老化の研究分野は混沌とした状況にある.このような混乱の原因を考察し,その対策を講じることが,細胞老化研究の真の発展に不可欠である.以下に,これまでの細胞老化研究の推移と,老化細胞の生体内での役割を解析する際の注意点を論じる.
1.細胞老化研究の推移
細胞老化(cellular senescence)は,ヒトの正常な組織から取り出した線維芽細胞をin vitroで継代培養すると,一定回数の細胞分裂を経て不可逆的に細胞分裂が停止する現象として発見された3).この現象は「分裂老化(replicative senescence)」ともよばれ,この原因としてさまざまな仮説が提唱されてきたが,今日ではヒトの線維芽細胞では細胞分裂に伴うテロメアの短縮が4),マウスの線維芽細胞の場合は酸化的ストレスが主な原因であることが明らかになっている5).一方,細胞老化は長らく培養細胞を用いて研究されてきたため,正常な体細胞をin vitroで培養したことによるストレスで引き起こされる現象,すなわちin vitro artifactである可能性も指摘されてきた6).しかし,その後,細胞老化を誘導する遺伝子の存在が示され7),p53,Rb,p21Waf1/Cip1/Sdi1やp16INK4aなどのがん抑制遺伝子が細胞老化の誘導に関与していることが明らかになり8)〜10),細胞老化はがん抑制機構として注目されるようになった(図).また,発がん性をもつ変異型ras遺伝子を正常な線維芽細胞に発現させると,p21Waf1/Cip1/Sdi1やp16INK4aの発現がすみやかに上昇し,細胞分裂が停止することが報告され11),細胞老化は単に細胞の分裂可能回数を制限しているだけでなく,もっと積極的に発がんリスクを低減させる役割を果たしていると認識されるようになった12).さらに,老化細胞にみられる特徴がヒトやマウスの前がん病変部で観察されることや13)〜15),p16INK4a遺伝子をノックアウトしたマウスでは発がんの頻度が著しく上昇すること16)等から,細胞老化はin vitro artifactではなく,生体内で重要ながん抑制機構として働いていることが広く認識されるようになった.また,活性酸素種(reactive oxygen species:ROS)が細胞老化の誘導と不可逆性の維持に寄与していることと17),p16INK4aノックアウトマウスでは,加齢に伴う組織幹細胞の自己複製能の低下が遅れることが相次いで報告され18)〜20),細胞老化ががん抑制機構として機能する一方で,組織幹細胞を枯渇させることで個体老化を促進している可能性が示唆されるようになった.実際,RT-qPCR21)やp16INK4a発現レポーターマウスを用いた解析により22),マウスにおいて加齢に伴い体内のさまざまな部位で老化細胞が蓄積することが確認されており,霊長類でも同様の結果が得られている23).これにより,細胞老化はがん抑制機構として機能する一方,個体老化を促進する側面も有していると考えられるようになった.
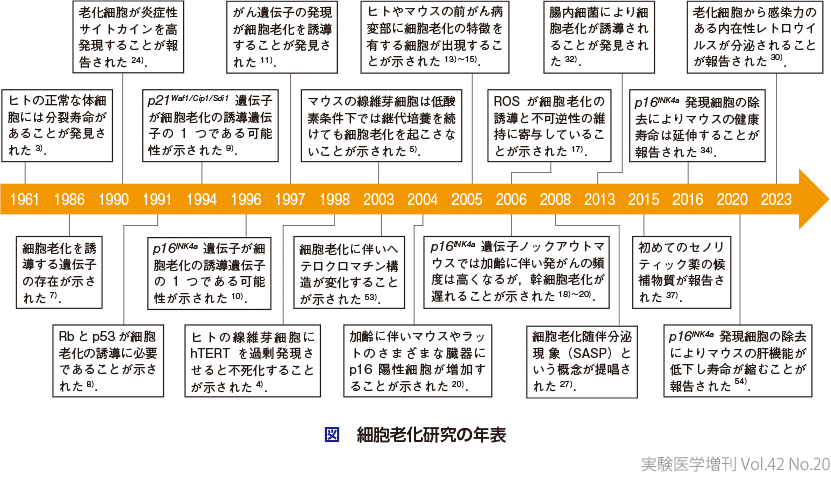
一方,古くから老化細胞は炎症性サイトカイン,ケモカイン,増殖因子,コラゲナーゼなどのさまざまな分泌因子を高発現することが報告されていた24)〜26).2008年,この現象にsenescence-associated secretory phenotype(SASP)という名称が付けられると27),老化細胞がSASPを介して周囲の細胞にさまざまな影響を及ぼす可能性があることが認識され,免疫学者を含む多くの研究者の注目を集めるようになった.SASPには分泌性タンパク質だけでなく,細胞外小胞(エクソソーム)28)29)や感染力を有する内在性レトロウイルスなども含まれており(第1章-4参照)30),多彩な機能をもつ可能性が指摘されている.実際,SASPはがんを含むさまざまな炎症性疾患の発症を促進する有害な作用をもつ一方31)32),がん抑制,組織修復,胎盤の機能維持,免疫機能の活性化といった生体に有益な役割を果たしている報告もある33).そのようななかで,Bakerらはp16INK4a遺伝子プロモーターの下流でアポトーシスを誘導できる遺伝子を発現するトランスジェニックマウス(INK-ATTACマウス)を作製し,p16INK4a遺伝子プロモーターが活性化した細胞を死滅させることで,マウス体内の老化細胞を減少させ,健康寿命を延ばせることを報告した34).この報告を機に,老化細胞の除去やSASPの制御が健康寿命の延伸につながるという考えが広まり,老化細胞を除去する薬剤(セノリティック薬)やSASPを制御する薬剤(セノモルフィック薬)の開発がさかんに行われるようになった35)〜37).
2.生体内での老化細胞の検出方法についての注意点
前述のように細胞老化はもともと,継代培養による分裂老化として認識されていたが,その後,がん遺伝子の発現や放射線,紫外線,DNA損傷性薬剤,酸化ストレス,炎症性サイトカインなど,さまざまなストレスによっても誘導されることが明らかになった12).また,細胞種やストレスの種類によって,遺伝子発現パターンやその他の表現型に多少違いが生じることもわかり38),生体内で老化細胞を特定することが難しくなってきている.そこで,最近,International Cell Senescence Association(国際細胞老化学会)を中心に,生体内で老化細胞を検出するための基準としてMinimal Information of Cellular Senescence Experimentation in vivo(MICSE)が提唱された39).具体的には,細胞種を問わず老化細胞に共通してみられる現象を複数調べ,さらに細胞種特異的な現象も考慮して判断することが推奨されている.以下に,老化細胞と判断するために調べておくべき共通事項を記す.
①生体内で細胞分裂が不可逆的に停止しているのか,それとも可逆的に停止しているのかを区別するのは困難である.しかし,少なくともEdUの取り込みがないことや,Ki-67などの細胞増殖関連遺伝子の発現が低下していることを調べ,細胞周期が停止していることは確認しておく.
②多くの老化細胞では,p16INK4aやp21Waf1/Cip1/Sdi1などのサイクリン依存性キナーゼ阻害因子(cyclin-dependent kinase inhibitor:CDKI)の発現が上昇する.そのため,まずこれらの発現レベルを確認する必要がある.ただし,細胞種によっては,p16INK4a以外のcdkn2遺伝子(p15INK4b,p18INK4c,p19INK4d)や,p21Waf1/Cip1/Sdi1以外のCip/Kip遺伝子(p27Kip1,p57Kip2)の発現上昇が細胞老化を引き起こす場合もある.また,CDKIの発現上昇がみられなくても,その下流で働くタンパク質(サイクリンやCDK)の発現レベルが低下し,Rbのリン酸化が阻害されるケースもあるため,これらの点も併せて調べておくべき.
③老化細胞では多くの場合,恒常的なDNA損傷応答(DNA damage response:DDR)の活性化がみられるため,DDRの代表的なマーカーであるγH2AX fociや53BP1 fociを免疫組織化学染色で確認する.また,細胞が単離可能であれば,コメットアッセイなどを用いて実際にDNA損傷があるかどうかも調べる方がよいだろう.
④老化細胞ではミトコンドリア機能の低下が頻繁にみられるため,ROSのレベルがコントロールとなる組織に比べて亢進しているかどうかを確認する.
⑤多くの老化細胞でSASPがみられるため,SASP因子の発現も調べる必要がある.ただし,SASP因子の種類や発現レベルは細胞種や生体内の状況によって異なるため,少数のSASP因子ではなく,可能な限り複数の因子を調べることが求められる.
関連して注意すべき点として,マウス組織におけるp16INK4aタンパク質の発現レベルはヒトの組織ほど高くない.そのため,抗体を用いてマウスの組織染色を行う際には,必ずp16INK4aノックアウトマウスの組織をネガティブコントロールとして使用すべきである.われわれとMayo ClinicのSundeep Khoslaのグループは,入手可能な多くのp16INK4a抗体を試した結果,ラビットモノクローナル抗体であるEPR20418がいくつかのマウスの非腫瘍組織で加齢に伴う内在性p16INK4aの発現上昇を検出できることを見出した40)41).ただし,組織染色においては,通常のPFA固定ではなくブアン固定を推奨する.発現レベルが非常に高い組織ではPFA固定でも検出可能だが,バックグラウンドシグナルが強くなるため,注意が必要である40).一方,最近では1細胞RNAシークエンシング(single-cell RNA sequencing:scRNA-seq)解析や空間トランスクリプトーム解析が可能となってきており,これらの解析方法を活用して内在性p16INK4a遺伝子を含む,細胞老化に伴い発現変化する複数の遺伝子の発現レベルを調べることが,生体内で老化細胞を検出するうえで最も効率的であると考えられる42).
3.老化細胞レポーターマウスについての注意点
これまで細胞老化反応をマウスの生体内で観察する目的や,老化細胞を体内から除去する目的でさまざまなマウスモデルが作製されてきた39).それらのほとんどは老化細胞で発現レベルが上昇することが知られているp21Waf1/Cip1/Sdi1,p16INK4a,p19ARF遺伝子のプロモーターを利用した遺伝子改変マウスである.表に示すようにそれら遺伝子のプロモーターの下流に発光遺伝子や蛍光遺伝子などのレポーター遺伝子や細胞死誘導遺伝子をつないだ遺伝子断片または染色体断片を導入したトランスジェニックマウスや,内在性のp16INK4a遺伝子座にレポーター遺伝子や細胞死誘導遺伝子を挿入したノックインマウスがほとんどである22)43)〜49).より忠実に内在性のp21Waf1/Cip1/Sdi1,p16INK4a,p19ARF遺伝子の発現を反映しやすいのはノックインマウスであると思われがちであるが,p16INK4a遺伝子の場合はプロモーター領域だけでなく50),イントロンやmRNAの3′非翻訳領域51)52),さらにはエピジェネティクス53)によっても発現レベルが制御されているためそれらの遺伝子構造を改変した形でノックインした場合はノックインマウスといえども,うまく内在性p16INK4aの発現を反映できない可能性があるので注意すべきである.また,ノックインマウスの場合は,p16INK4aの機能が阻害されている可能性があることも認識しておくべきであろう.

最近はCre54)またはCre-ERT255)をp16INK4aやp21Waf1/Cip1/Sdi1遺伝子のプロモーター領域の下流にノックインしたマウスとCreの発現により蛍光タンパク質や細胞死誘導遺伝子を発現するようにした遺伝子改変マウスとを交配することでp16INK4aやp21Waf1/Cip1/Sdi1の発現をモニターしたり,それら遺伝子を発現する細胞を死滅させることが可能なマウスが報告されている(第4章-2を参照).特にp16INK4a遺伝子の場合,プロモーター活性が低いのでCreにより間接的にレポーターや細胞死誘導遺伝子を発現させることでシグナルを増強しうるというメリットがある.しかし,これらのマウスはいったんp16INK4aまたはp21Waf1/Cip1/Sdi1プロモーターが活性化したら,その後プロモーター活性が低下しても,レポーターが発現し続けるため,必ずしもp16INK4aやp21Waf1/Cip1/Sdi1遺伝子の発現をリアルタイムにモニターしているわけではないことに留意すべきである.さらに注意すべき点は遺伝子改変する際に用いたneomycin耐性遺伝子発現ユニット(neomycinカセット)が取り除いてあるかどうかである.neomycinカセットを残したままにしておくとneomycinカセットに組込まれている強力なプロモーターが活性が弱い内在性p16INK4a遺伝子プロモーターに影響を及ぼしてしまい,内在性p16INK4a遺伝子の発現を反映できなくなる可能性が指摘されている39).この違いがOmoriら55)のマウス(neomycinカセットが残っている)とGrosseら54)のマウス(neomycinカセットが取り除いてある)の表現型の違いの一因になっている可能性がある.いずれにせよ,レポーターの発現が内在性p16INK4a遺伝子の発現をうまく反映しているかどうかをscRNA-seq解析で確認をしておくことが必要である.内在性p16INK4a遺伝子やp21Waf1/Cip1/Sdi1遺伝子の発現を反映していないなど,問題があるマウスを使用してしまうと,間違った結果を論文にしてしまい,科学をゆがめてしまう危険性がある.そのような危険性を回避するためには,報告された老化細胞レポーターマウスのデータを鵜呑みにせず,それらのマウスを入手したら,まずは各自でそのマウスが本当に機能するかどうかを確認してから実験を開始されることを推奨する.以下,われわれがp16-3MRマウスで経験した問題について紹介する.
p16-3MRマウスはマウスのp16INK4a遺伝子座約50 kbの中のp16INK4a遺伝子プロモーターの下流に3MR遺伝子〔Renillaのluciferase(Rluc)とRFPに加えヘルペスウイルスのチミジンキナーゼ(HSV-TK)をタンデムにつないだ融合タンパク質を発現する人工遺伝子〕を組込んだ染色体断片をもつトランスジェニックマウスである56).このため,p16INK4a遺伝子の発現をRlucによる生物発光シグナルとRFPによる蛍光シグナルで検出でき,ガンシクロビル(GCV)の投与でp16INK4a発現細胞を死滅させることが可能なマウスとしてCampisi博士のラボから発表された56).その後このマウスは世界中で使われるようになり,その成果がNature誌やCell誌等の一流誌に掲載されている57)58).しかし,その一方で,p16-3MRマウスはうまく機能しないという噂を耳にするようになった.筆者自身はp16-3MRマウスを使った実験を担当していたわけではないが,Demaria et al(2014)56)の共著者であったことから,責任を感じて2016年にp16-3MRマウスをCampisi研究室から入手し,7年以上の歳月をかけ慎重に検証した.その結果,p16-3MRマウスではトランスジーン(3MR遺伝子)の発現レベルはきわめて低く,Demaria et al.(2014)56)で報告されたデータを再現することはできなかった59).なぜこのようなマウスが論文として報告されてしまったのかだが,その主な原因はネガティブコントロール(野生型マウス)が用いられていなかったことと,p16-3MRマウスは黒毛マウスであるにもかかわらず,毛を剃らずに生物発光イメージングを行っていたことにあると考えられる.Rlucの発光基質であるCoelenterazineは血清アルブミンやROSと反応すると,Rlucの発現とは無関係に弱く発光することが知られている60)61).このバックグラウンドシグナルが加齢や放射線照射等によりマウスの体毛が抜けたり白髪になったために検出されやすくなったことを,Rlucの発現レベルが上昇したためだと勘違いしたことにあると思われる.これだけでは説明できないデータもDemaria et al.(2014)には多数含まれているが56),いずれにせよ,Campisi研究室のような有名な研究室で開発されたマウスなら間違いがないだろうという思い込みで誤ったデータが学術論文として発表されてしまった可能性も考えられる.私の共著の論文がこのような問題を引き起こしてしまったことに大変申しわけなく思うと同時に,有名ラボから発表されたマウスモデルといえど決して鵜呑みにせず,検証してから使用されることを推奨する.
4.老化細胞除去(セノリシス)についての注意点
BakerらによりINK-ATTACマウスを用いてp16INK4a遺伝子プロモーターが活性化した細胞を死滅させることで,マウスの健康寿命が延びることが報告されて以来34),セノリシスが注目され,セノリティック薬の開発がさかんに行われている36).現在までに20種類以上の薬剤がセノリティック薬として報告され37),なかにはマウスに投与することで健康寿命の延長効果がみられることが報告されているものも存在する62)63).しかし,現在までのところ,臨床試験で統計学上有意な効果が認められている薬剤は見つかっていない64).われわれは他の研究室と協力して現在までに報告のある入手可能なセノリティック薬のほとんどを用いて比較実験を行ったが,老化細胞を特異的に死滅させるセノリティック薬はごく一部で,多くの薬剤が老化細胞を死滅させる濃度ではコントロール細胞(非老化細胞)も死滅させたり,増殖を阻害したりすることを見出している(第3章-3参照).もちろん単にわれわれが正しく実験条件を再現できていない可能性を完全に排除することはできないが,それにしても何かがおかしいと感じざるを得ない.例えば,免疫チェックポイント阻害薬であるPD-1抗体を老齢マウスに投与することで体内の老化細胞を減らし,健康状態を改善させることが報告され65),テレビや週刊誌等でもさかんに報道されている.しかし,最近別のグループからPD-1抗体をマウスに投与して免疫チェックポイントを阻害しても老化細胞の減少が確認されないし,健康状態も改善されないことが報告されている66).文献66では抗PD-1抗体と抗PD-L1抗体それぞれの効果をみていて,抗PD-L1抗体のみで老化細胞の有意な減少を確認している.注目すべきことは,この論文では抗PD-L1抗体の投与でp16とPD-L1を共発現した細胞が減るのはPD-L1に対するアゴニスト抗体を投与した場合であり,免疫チェックポイント機構を阻害するPD-L1に対するアンタゴニスト抗体を投与した場合ではないことが明記されている.すなわち免疫チェックポイント機構を阻害しても老化細胞は減少しないことになる.
ではなぜセノリティック薬に関するデータはこのように再現性が取りにくいのであろうか?さまざまな可能性が考えられるが,1つの可能性として,マウスは高齢になると個体差が大きくなることにあるのかもしれない.このため,用いるマウスの数をかなり増やして実験を行わないと誤った結論を導き出してしまう可能性がある.例えば,フラボノイドの一種であるFisetinにはセノリシス活性があり,85週齢の高齢マウスに投与すると寿命延長効果が現れることが報告されているが,その論文で使用されたマウスの数は各群8~9匹である62).一方,最近,別のグループがその30倍ほどの数のマウスを用いて追試を行ったところ,Fisetinに寿命延長効果は確認できないことが報告されている67).もちろんこの違いは飼育条件やマウスの系統が微妙に異なるせいである可能性も排除できないが,一般的には多数のマウスを用いた実験結果の方が信頼性が高く少数のマウスを用いた実験結果を信用するのはリスクが高いように感じる.寿命延長効果など社会の注目度が高いトピックスに関しての研究結果を報告する際にはより慎重に研究結果を検証したうえで論文として発表すべきだと筆者は感じる.個体差を考慮して実験に用いるマウスの数を十分増やすことと,研究者の恣意的な要素が入らないようにブラインドで(解析担当者がどの薬剤が投与されたマウスを解析しているのかわからないようにして)実験を行うことを強く推奨する.
おわりに
細胞老化の研究が社会の注目を集めるようになったことで,研究者の数や関連するスタートアップ企業の数が急速に増え,細胞老化に関する研究が加速しつつあること自体は望ましいことではあるが,実験結果の誇張や歪曲,再現性の欠如など慎重さを欠く論文が散見されることは大きな問題である.このような状況が放置されると,科学に対する社会の信頼を失いかねない.特集の序論としては少しそぐわない内容かもしれないが,長年細胞老化の研究に携わってきた者として68),ここに懸念を表明する.本書では,細胞老化関連研究の最前線を行く研究者の方々に,冷静かつ慎重に最新の研究成果を概説していただいた.しかし,誌面スペースの都合上,すべての研究者に依頼できなかったことをお詫び申し上げる.本書が,細胞老化研究が冷静さを取り戻し,真に発展する一助となることを願う.
文献
- Rodier F & Campisi J:J Cell Biol, 192:547-556, doi:10.1083/jcb.201009094(2011)
- Watanabe S, et al:Cancer Sci, 108:563-569, doi:10.1111/cas.13184(2017)
- Hayflick L & Moorhead PS:Exp Cell Res, 25:585-621, doi:10.1016/0014-4827(61)90192-6(1961)
- Bodnar AG, et al:Science, 279:349-352, doi:10.1126/science.279.5349.349(1998)
- Parrinello S, et al:Nat Cell Biol, 5:741-747, doi:10.1038/ncb1024(2003)
- Sherr CJ & DePinho RA:Cell, 102:407-410, doi:10.1016/s0092-8674(00)00046-5(2000)
- Lumpkin CK Jr, et al:Science, 232:393-395, doi:10.1126/science.2421407(1986)
- Shay JW, et al:Exp Cell Res, 196:33-39, doi:10.1016/0014-4827(91)90453-2(1991)
- Noda A, et al:Exp Cell Res, 211:90-98, doi:10.1006/excr.1994.1063(1994)
- Hara E, et al:Mol Cell Biol, 16:859-867, doi:10.1128/MCB.16.3.859(1996)
- Serrano M, et al:Cell, 88:593-602, doi:10.1016/s0092-8674(00)81902-9(1997)
- Serrano M & Blasco MA:Curr Opin Cell Biol, 13:748-753, doi:10.1016/s0955-0674(00)00278-7(2001)
- Collado M, et al:Nature, 436:642, doi:10.1038/436642a(2005)
- Michaloglou C, et al:Nature, 436:720-724, doi:10.1038/nature03890(2005)
- Braig M, et al:Nature, 436:660-665, doi:10.1038/nature03841(2005)
- Sharpless NE, et al:Oncogene, 23:379-385, doi:10.1038/sj.onc.1207074(2004)
- Takahashi A, et al:Nat Cell Biol, 8:1291-1297, doi:10.1038/ncb1491(2006)
- Janzen V, et al:Nature, 443:421-426, doi:10.1038/nature05159(2006)
- Krishnamurthy J, et al:Nature, 443:453-457, doi:10.1038/nature05092(2006)
- Molofsky AV, et al:Nature, 443:448-452, doi:10.1038/nature05091(2006)
- Krishnamurthy J, et al:J Clin Invest, 114:1299-1307, doi:10.1172/JCI22475(2004)
- Yamakoshi K, et al:J Cell Biol, 186:393-407, doi:10.1083/jcb.200904105(2009)
- Herbig U, et al:Science, 311:1257, doi:10.1126/science.1122446(2006)
- Bauer EA, et al:Exp Cell Res, 161:484-494, doi:10.1016/0014-4827(85)90103-x(1985)
- Maier JA, et al:Science, 249:1570-1574, doi:10.1126/science.2218499(1990)
- Tahara H, et al:Oncogene, 11:1125-1132, doi:undefined(1995)
- Coppé JP, et al:PLoS Biol, 6:2853-2868, doi:10.1371/journal.pbio.0060301(2008)
- Takahashi A, et al:Nat Commun, 8:15287, doi:10.1038/ncomms15287(2017)
- Takasugi M, et al:Nat Commun, 8:15729, doi:10.1038/ncomms15728(2017)
- Liu X, et al:Cell, 186:287-304.e26, doi:10.1016/j.cell.2022.12.017(2023)
- Chan ASL & Narita M:Genes Dev, 33:127-143, doi:10.1101/gad.320937.118(2019)
- Yoshimoto S, et al:Nature, 499:97-101, doi:10.1038/nature12347(2013)
- de Magalhães JP:Science, 384:1300-1301, doi:10.1126/science.adj7050(2024)
- Baker DJ, et al:Nature, 530:184-189, doi:10.1038/nature16932(2016)
- Zhu Y, et al:Aging Cell, 14:644-658, doi:10.1111/acel.12344(2015)
- Chaib S, et al:Nat Med, 28:1556-1568, doi:10.1038/s41591-022-01923-y(2022)
- Power H, et al:Aging Cell, 22:e13948, doi:10.1111/acel.13948(2023)
- Wechter N, et al:Aging (Albany NY), 15:2824-2851, doi:10.18632/aging.204666(2023)
- Ogrodnik M, et al:Cell, 187:4150-4175, doi:10.1016/j.cell.2024.05.059(2024)
- Kawamoto S, et al:Nat Cell Biol, 25:865-876, doi:10.1038/s41556-023-01145-5(2023)
- Doolittle ML, et al:Nat Commun, 14:4587, doi:10.1038/s41467-023-40393-9(2023)
- Gurkar AU, et al:Nat Aging, 3:776-790, doi:10.1038/s43587-023-00446-6(2023)
- Ohtani N, et al:Proc Natl Acad Sci U S A, 104:15034-15039, doi:10.1073/pnas.0706949104(2007)
- Hashimoto M, et al:JCI Insight, 1:e87732, doi:10.1172/jci.insight.87732(2016)
- Burd CE, et al:Cell, 152:340-351, doi:10.1016/j.cell.2012.12.010(2013)
- Baker DJ, et al:Nature, 479:232-236, doi:10.1038/nature10600(2011)
- Liu JY, et al:Proc Natl Acad Sci U S A, 116:2603-2611, doi:10.1073/pnas.1818313116(2019)
- Reyes NS, et al:Science, 378:192-201, doi:10.1126/science.abf3326(2022)
- Haston S, et al:Cancer Cell, 41:1242-1260.e6, doi:10.1016/j.ccell.2023.05.004(2023)
- Ohtani N, et al:Nature, 409:1067-1070, doi:10.1038/35059131(2001)
- Bracken AP, et al:Genes Dev, 21:525-530, doi:10.1101/gad.415507(2007)
- Kotake Y, et al:Genes Dev, 21:49-54, doi:10.1101/gad.1499407(2007)
- Narita M, et al:Cell, 113:703-716, doi:10.1016/s0092-8674(03)00401-x(2003)
- Grosse L, et al:Cell Metab, 32:87-99.e6, doi:10.1016/j.cmet.2020.05.002(2020)
- Omori S, et al:Cell Metab, 32:814-828.e6, doi:10.1016/j.cmet.2020.09.006(2020)
- Demaria M, et al:Dev Cell, 31:722-733, doi:10.1016/j.devcel.2014.11.012(2014)
- Moiseeva V, et al:Nature, 613:169-178, doi:10.1038/s41586-022-05535-x(2023)
- Baar MP, et al:Cell, 169:132-147.e16, doi:10.1016/j.cell.2017.02.031(2017)
- Hori N, et al:bioRxiv, doi:10.1101/2024.06.24.600181(2024)
- Zhao H, et al:Mol Imaging, 3:43-54, doi:10.1162/15353500200403181(2004)
- Bronsart LL, et al:PLoS One, 11:e0146601, doi:10.1371/journal.pone.0146601(2016)
- Yousefzadeh MJ, et al:EBioMedicine, 36:18-28, doi:10.1016/j.ebiom.2018.09.015(2018)
- Xu Q, et al:Nat Metab, 3:1706-1726, doi:10.1038/s42255-021-00491-8(2021)
- Farr JN, et al:Nat Med, 30:2605-2612, doi:10.1038/s41591-024-03096-2(2024)
- Wang TW, et al:Nature, 611:358-364, doi:10.1038/s41586-022-05388-4(2022)
- Majewska J, et al:Nat Cell Biol, 26:1336-1345, doi:10.1038/s41556-024-01465-0(2024)
- Harrison DE, et al:Geroscience, 46:795-816, doi:10.1007/s11357-023-01011-0(2024)
- Hara E:Nat Cell Biol, 26:176, doi:10.1038/s41556-023-01306-6(2024)
<著者プロフィール>
原 英二:東京理科大学大学院修了,1993年(米国)University of California, Berkeleyポスドク,’95年(英国) Imperial Cancer Research Fund Laboratoriesポスドクを経て’98年に(英国)Cancer Research UK, Paterson Institute for Cancer Researchでグループリーダーとして研究室を主宰,2003年徳島大学ゲノム機能研究センター教授,’08年公益財団法人がん研究会がん研究所部長,’15年より大阪大学微生物病研究所教授(免疫学フロンティア研究センターを兼任).大学院生の頃より一貫して細胞老化の研究を行っている.

