
第1章 概論と基礎知識
2 AlphaFold2の衝撃
大上雅史(東京科学大学情報理工学院)
構造バイオインフォマティクスのなかでも最も重要視されてきたタンパク質立体構造予測問題.AlphaFold2の登場によって立体構造予測はどのように変化したのか.AlphaFold2の登場前夜から今日に至るまでの動向を,周辺技術も含めて俯瞰しつつ,今後の展望について紹介する.
はじめに
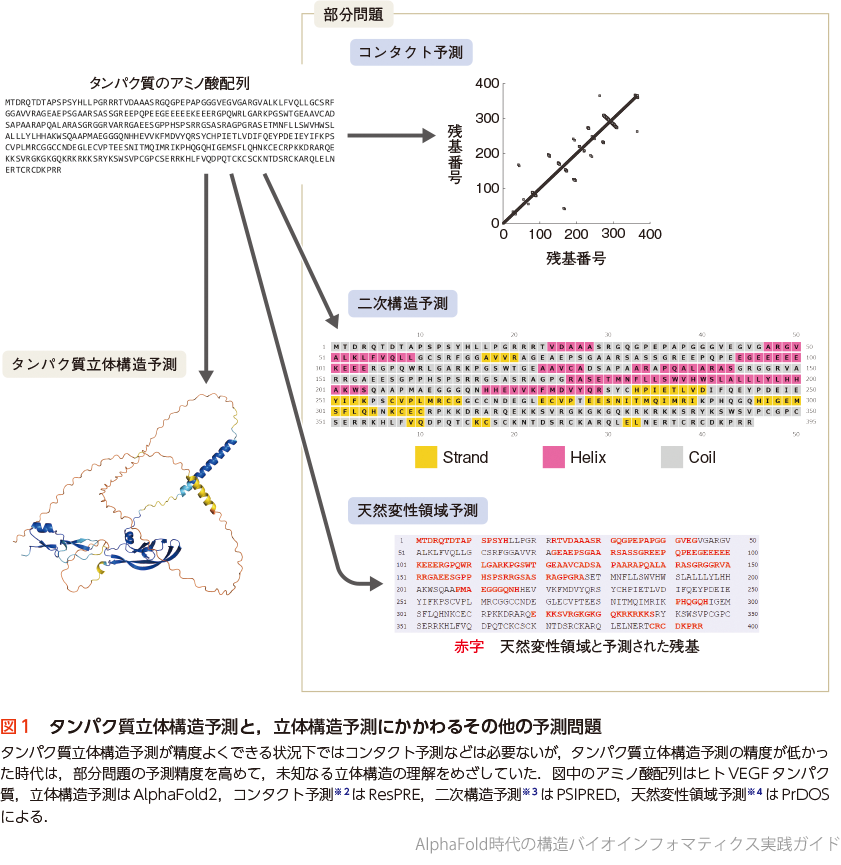
構造バイオインフォマティクスの最も主要な問題の一つに,タンパク質のアミノ酸配列から立体構造を予測する「タンパク質立体構造予測」がある(図1).構造生物学によってタンパク質の立体構造(原子の三次元座標)を1つひとつ実験的に決定してきた経緯からもわかるように,タンパク質の立体構造を解くことは生命科学分野において,また医薬品設計などの分野においても大きな意味をもつ.この構造生物学の発展と並行して,タンパク質の立体構造を高い実験コストを費やさずになんとか配列から推測することはできないかと,情報科学技術による予測の研究が進められていた.1994年にはじまったタンパク質立体構造予測の技術を競う国際コンペティションCASP※1が隔年で開催されるなかで,数多くのタンパク質立体構造予測手法が提案され,研究者達がしのぎを削ってきた.2021年に発表されたAlphaFold21)も,CASPが生んだ多数の予測プログラムの1つである.
AlphaFold2 の登場
AlphaFold2について,「あたかも突然降って湧いたAIによってタンパク質立体構造予測問題が解決されてしまった」かのように語られることも多い.しかし,タンパク質立体構造予測は生命科学における重要な問題として古くから認識され,さまざまな研究がなされてきた.現にAlphaFold2を開発したGoogle DeepMind社は,AlphaFold2の前身にあたるAlphaFold(AlphaFold1)2)を2019年に発表しており,またコンタクト予測※2や二次構造予測※3,天然変性領域予測※4といったタンパク質立体構造予測にかかわる主要タスク(図1)にいたっては,2010年代前半の時点ですでに深層学習(deep learning)による研究が進められていた3)4).AlphaFold2は,AI・機械学習の技術の進展,タンパク質立体構造データの蓄積,計算機自体の発展に,さらにCASPを通じて蓄積されたさまざまな生物物理学的知見が幾層にも積み重なって結集した成果なのである.
AlphaFold2の性能
1.CASP14の結果および論文による報告
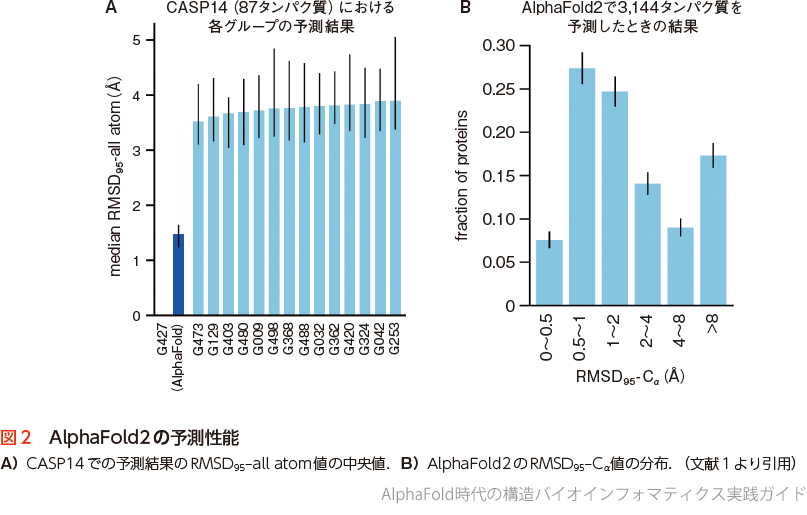
2020年に開催されたCASP14の報告で,AlphaFold2の予測は正解構造との差異を表すglobal distance test–total score(GDT_TS)※5の中央値で92.4という値を叩き出した.GDT_TSは90を超えると実験的に得られる構造と同等レベルの予測とみなすことができ,Science誌ではゲームチェンジャーと評されるほど,その予測精度は従来の構造予測法を大きく上回るものであった.
図2は文献1で報告されたAlphaFold2の予測性能のサマリーである.予測構造と実験的に得られた構造との差異を示すRMSD95値※6で評価されている.図2Aが他のグループとAlphaFold2のCASP14での成績の比較であり,出題された87タンパク質に対するRMSD95値の中央値が示されている.他のグループが>3 Åの予測結果となるなかで,AlphaFoldグループは約1.5 Åの良好な予測結果であったことが報告された.また,図2BはCASP14以外のタンパク質で大規模にAlphaFold2の予測を行った結果であり,半数以上のタンパク質でRMSD95–Cα <2.0 Åとなる予測が得られたことが示されている.
2.具体的な予測構造の例
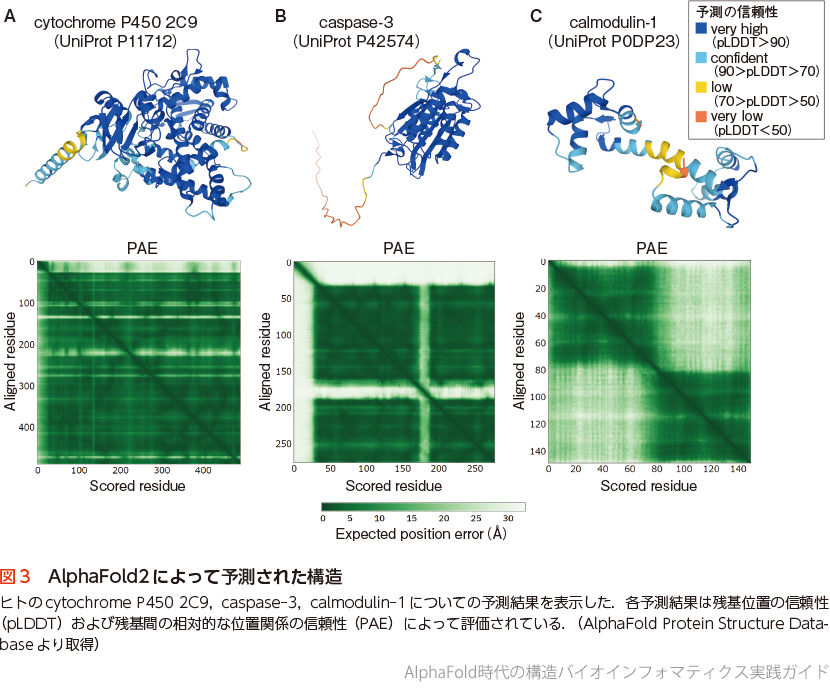
AlphaFold2の予測構造を取得するには,自身でAlphaFold2を実行する以外に,AlphaFold Protein Structure Database(AlphaFold DB)5)から予測済みの構造を参照することもできる.図3にAlphaFold DBで検索した3例の予測構造を示した.各予測結果は,pLDDT(predicted local distance difference test)とよばれる各残基位置の予測の信頼性指標と,PAE(predicted aligned error)とよばれる残基間の相対位置関係の信頼性指標とともに提供され,ユーザーはこれらの指標をもとに予測構造の妥当性をある程度判断することができる.例えばcytochrome P450 2C9タンパク質の予測結果は全長にわたって高いpLDDT値となっており,またPAEも全体的に良好であることからおおむね信頼できる予測構造であると判断できる(図3A).caspase-3では主要な領域については高いpLDDT値となっているが,1〜31番残基と169〜190番残基についてはpLDDT値が低くなっており,またPAEについても薄くなっているため,これらの領域については構造をそのまま信用することは危険である(図3B).calmodulin-1については中間にあるヘリックスの構造のpLDDTが低く,さらにPAEが特徴的な結果となっている(図3C).この例のようにPAEが市松模様のような結果となるケースは,マルチドメインタンパク質に多くみられ,各ドメインの予測構造の信頼性は高いものの,ドメイン間の位置関係については正しくない可能性があるときに起きる.実際のところcalmodulin-1はダンベル型タンパク質として知られており,Ca2+イオンの結合によって大きく構造変化を起こすことがわかっている.
3.よくない予測結果となるケース
文献1や図2Bで報告されている通り,どのようなケースにおいてもAlphaFold2が完璧な予測を返すわけではない.特に図2Bのグラフにあるように,RMSDが8.0 Åを超えるケースが約17%存在することが示されている.実際に図3のcaspase-3やcalmodulin-1でも予測結果の信頼性が低い可能性について言及したが,pLDDTやPAEなどをもとに予測結果を実際に利用するかどうかを判断することが重要となる.以下に具体例を示す.
- 図3Bのcaspase-3のように配列の末端はpLDDTが低くなりやすい.実際に末端は実験的に決定された構造でも座標が登録されていないことも多いため,注意が必要である.
- 図3Bのcaspase-3のように,途中の領域でpLDDTが低くなるループ領域がみられることがある.この領域はおそらく特定の構造をとらない天然変性領域と考えられる.実際にcaspase-3はPDBに多くの構造が存在するが,この領域の残基はunmodeled residueとして座標がない状態で登録されているものがほとんどである.
- 図3Cのcalmodulin-1のように,ドメイン間の位置関係が信頼できないケースが存在する.特にcalmodulin-1はCa2+や他のタンパク質/ペプチドとの結合によって大きく構造変化を起こすことが知られており,PDBにも多様な構造が登録されている.calmodulin-1のようにさまざまなコンフォメーションの可能性があるタンパク質において,AlphaFold2の予測はそのなかのどの状態のコンフォメーションに近い構造になるかがわからないため注意が必要である(逆にAlphaFold2のパラメータを操作するなどして別のコンフォメーションを出力する試みもある6)).
- AlphaFold2は入力された配列に対する類似配列検索の結果によって大きく予測精度が変わることが知られており,具体的には類縁配列がおよそ100本未満となるタンパク質の予測性能は低くなる可能性がある1).AlphaFold DBでは配列検索の経過に関する情報を見ることができないが,AlphaFold2本体や次項に示すColabFoldでは配列検索の結果も参照することができるので,類縁配列が少なくないかを確認することが推奨される.
ColabFoldの貢献
AlphaFold2以前のAlphaFold1も,当時のタンパク質立体構造予測技術としては世界最高精度を達成していた.にもかかわらずAlphaFold1は世の中にほとんど浸透しなかった.じつはAlphaFold1はプログラムが一部しか公開されなかったために,ユーザーが公開されている部分を拾って使っても,立体構造予測を行うことができなかったのである*1.
AlphaFold2は公開当初から,予測構造の出力までを一貫して可能にした実行コードが提供され,ユーザーは実際にインストールして実行することができた.実際,公開直後から実行結果に関するtwitter(現X)でのつぶやきが多々なされ,その予測精度に驚く声も多かった.一方で,数TB(テラバイト)のデータベースを保存できるGPU搭載のLinuxマシンが要求されたため,必ずしも「気軽」に試せるというわけではなかった.しかしすぐに2つの解決策がGoogle DeepMind社より提供された.1つはあらかじめ別途に予測した構造を使えるようにするというアプローチであり,これは先に紹介したAlphaFold DBとして提供されている.もう1つはGoogle Colaboratoryを介したクラウド実行環境の提供であり,ユーザーはブラウザ画面上で配列を入力するだけで,あとはほぼ1クリックで予測の実行ができるようになった.
ただ,当初Google DeepMind社から公開されたGoogle Colaboratoryの実行環境は,いわゆる「簡易版」であり,AlphaFold2本来の予測性能からは劣るものであった.その後,Google DeepMind社とは別の研究グループから発表されたColabFold7)は,Google Colaboratoryのユーザビリティをそのままに,感度のよい高速な配列検索サーバーの採用8)などの工夫で,AlphaFold2本来の予測性能と同等性能を担保しつつ,20倍前後の予測計算時間の高速化に成功している.現在ではColabFoldも広く活用されており,「AlphaFold2を利用した」という文脈でColabFoldが使われていることも多い(内容の詳細や利用シーンについては第2章–3に譲る).
AlphaFold2のなかみ
基本的に,配列が似ているタンパク質は立体構造も似ていることが経験的にわかっており,もし立体構造が既知のタンパク質と40%以上の配列の一致があれば,その構造をテンプレートとしたホモロジーモデリング法※7によっておおむね正しい構造を推定できる.しかし,AlphaFold2は明確なテンプレートがなくてもよい予測結果を返す.なぜこのような高い予測性能を発揮したのか,そこには大きく3つのエッセンスがあると考えられる(図4).
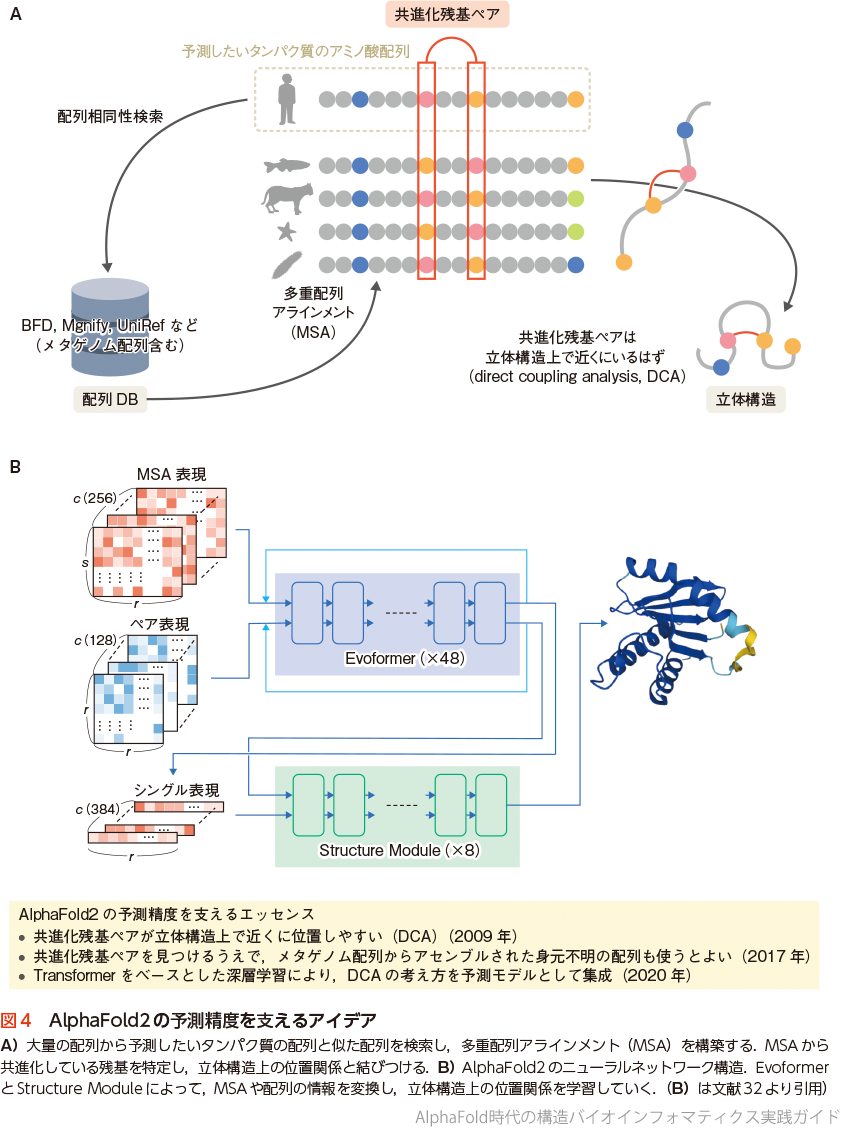
第一に,direct coupling analysis(DCA)とよばれる手法の採用がある.タンパク質の進化関係を考えたときに,タンパク質の各アミノ酸はそれぞれが勝手に(独立に)進化しているわけではなく,立体構造上で近い部位のアミノ酸同士は互いに依存して「共進化」しているという考え方および統計情報の利用である.この逆を考えれば,互いに依存している共進化残基ペアがわかれば,その残基は立体構造上のコンタクト残基となっていることが期待できる.共進化残基ペアは,配列データベースから進化的に近い配列をBLASTなどで検索し,多重配列アラインメント(multiple sequence alignment:MSA)を構築することで検出できる.このDCAの考え方は2009年にコンタクト予測の枠組みのなかで提案され9),2011年には立体構造予測に応用されはじめた10).
第二に,タンパク質の進化関係の知識を網羅するために,とにかく大量のタンパク質配列を活用するというアイデアがある.タンパク質の配列データベースであるUniProtを用いることはもちろんのこと,環境中の微生物叢からシーケンシングされた「生物種どころかどんなタンパク質かもわからないごたまぜ状態の」配列(メタゲノム配列)も併用して構築されたMSAを使うことで,構造予測精度を向上させるアイデアが2017年に提案された11).このとき,メタゲノム配列はゲノムアセンブリ法によってタンパク質と考えられるかたまり(コンティグ)になるまで繋ぎ合わせた配列が用いられる.
第三に,近年の機械学習技術の成果である深層学習の利用である.自然言語処理分野で発展したTransformer12)とよばれるニューラルネットワークを,MSAからの知識抽出(Evoformer)とMSA情報からの立体構造復元(Structure Module)にそれぞれ利用することで,DCAの考え方に基づくタンパク質立体構造予測を実現した(図4B).特筆すべきは,入力のタンパク質配列から出力の予測立体構造までを一気通貫に(end-to-endに)予測させることに成功しており,この点はコンタクト予測までを深層学習にゆだねていたAlphaFold1からの大幅なアップデートでもある.
ご覧ください
文献
- Jumper J, et al:Nature, 596:583-589, doi:10.1038/s41586-021-03819-2(2021)
- Senior AW, et al:Nature, 577:706-710, doi:10.1038/s41586-019-1923-7(2020)
- Lena PD, et al:Bioinformatics, 28:2449-2457, doi:10.1093/bioinformatics/bts475(2012)
- Eickholt J & Cheng J:BMC Bioinformatics, 14:88, doi:10.1186/1471-2105-14-88(2013)
- Tunyasuvunakool K, et al:Nature, 596:590-596, doi:10.1038/s41586-021-03828-1(2021)
- Sala D, et al:Curr Opin Struct Biol, 81:102645, doi:10.1016/j.sbi.2023.102645(2023)
- Mirdita M, et al:Nat Methods, 19:679-682, doi:10.1038/s41592-022-01488-1(2022)
- Steinegger M & Söding J:Nat Biotechnol, 35:1026-1028, doi:10.1038/nbt.3988(2017)
- Weigt M, et al:Proc Natl Acad Sci U S A, 106:67-72, doi:10.1073/pnas.0805923106(2009)
- Marks DS, et al:PLoS One, 6:e28766, doi:10.1371/journal.pone.0028766(2011)
- Ovchinnikov S, et al:Science, 355:294-298, doi:10.1126/science.aah4043(2017)
- Vaswani A, et al:Advances in Neural Information Processing Systems, 30:6000-6010(2017)
参考図書
- 大上雅史/企画:いま知りたい!! 使ってわかったAlphaFoldのリアル.「実験医学2022年2月号」, pp423-438, 羊土社(2022)
- 「実験医学2023年10月号 AlphaFoldの可能性と挑戦」(富井健太郎/企画),羊土社(2023)
- 福永津嵩:逆イジング法の生命情報データ解析への応用.JSBi Bioinformatics Review, 1:3-11, doi:10.11234/jsbibr.2020.1(2020)
- 富井健太郎:AlphaFoldによる蛋白質立体構造予測から機能予測へ.生物物理,64:5-11, doi:10.2142/biophys.64.5(2024)
- 柳澤渓甫:タンパク質立体構造情報を用いた薬剤バーチャルスクリーニング.JSBi Bioinformatics Review, 2:76-86, doi:10.11234/jsbibr.2021.9(2021)
- 山口秀輝,齋藤 裕:タンパク質の言語モデル.JSBi Bioinformatics Review, 4:52-67, doi:10.11234/jsbibr.2023.1(2023)
- 森脇由隆:AlphaFold2までのタンパク質立体構造予測の軌跡とこれから.JSBi Bioinformatics Review, 3:47-60, doi:10.11234/jsbibr.2022.3(2022)



