概論
体細胞モザイクとクローン進化(拡大と陽性選択)の概念
Outline of “somatic mosaicism”and“clonal evolution through positive
selection and expansion”
垣内伸之
Nobuyuki Kakiuchi1)2):The Hakubi Center for Advanced
Research, Kyoto University1)/Department of Gastroenterology and
Hepatology, Graduate School of Medicine, Kyoto
University2)(京都大学白眉センター1)/京都大学大学院医学研究科消化器内科学2))
次世代シークエンサーの登場に端を発してゲノム解析技術が飛躍的に向上し,がんの起源を探索する過程で正常細胞における遺伝子変異や正常組織における変異クローン拡大についての詳細が解明されつつある.個体を構成する細胞は絶えず分裂し発生・成長過程では総数が増加する一方,成人期以降はおおむね一定数となるが,陽性選択された変異クローンは増殖・拡大してしばしば臓器全体が変異クローンによって再構築されるに至る.本稿では体細胞モザイクとクローン進化について,その概念をいくつかの例を挙げて解説する.
はじめに
われわれヒトの個体は,1個の受精卵を起源として細胞分裂がくり返され,成人では約40兆個の細胞から構成される.細胞分裂の際にDNAはきわめて正確に複製されるが,正常細胞であっても1分裂あたり1~10個/30億塩基の割合で新たに変異が生じることがわかってきた1).すなわち,分裂後の娘細胞は互いに,もしくは,由来した細胞とはゲノムがわずかに異なっており,このことは,変異に基づいて,受精卵を頂点とするすべての体細胞の系統樹が作成可能であることを意味する.がんはその起源として一つの祖先細胞に由来し,ドライバー変異を獲得したクローンが増殖・拡大した結果として生じると考えられている.近年のゲノム解析技術の向上により,がんの網羅的ゲノム解析がなされた結果,多数のドライバー遺伝子が同定された2).一つの腫瘍が複数のドライバー変異を有することから,ドライバー変異の獲得は多段階にわたってなされると想定されるが,がんゲノム解析からは発がん過程の初期においてどのような細胞に,いつ,どのような変異が生じ,どのようなメカニズムによって細胞が自然選択されるかについての詳細は長らく不明であった.
最近,形態学的に正常な組織においても,変異を獲得したクローンが陽性に選択され,クローンが増殖・拡大していることが報告された3).正常組織で同定される変異の多くは各臓器に由来するがんと共通することが多いため,これら正常組織におけるクローン拡大はがんの起源である可能性が示唆されている.また,しばしばこれらの変異クローンによって組織の大部分が置換されることがあり,正常組織におけるクローン拡大は発がんのみならず,加齢や慢性炎症性疾患などとどのように関連しているのかを理解することは重要な課題となっている.本稿では正常組織におけるクローン拡大の最新の知見について述べる.
1体細胞モザイクとクローン拡大
❶ 正常組織における体細胞変異の獲得
がんの起源を探る研究として,古くはX染色体不活性化(lyonization)が用いられた.女性の細胞において父由来もしくは母由来のどちらか片方のX染色体は胎生期にランダムに不活性化されることを利用し,がんにおいてこの不活性化が片側アレルに偏ることから,がんは単一の細胞を起源とすることが明らかになった4).同様の解析を血液細胞に対して行うと,健常者であっても高齢者でこの偏り(skewed chromosome X inactivation)がしばしば観察され,そのような高齢者はその後の血液悪性腫瘍の発症リスクが上昇していることが明らかとなり,正常組織におけるクローン拡大が観察された最初の例となった5).X染色体の不活性化は胎生期に完了することに比べて,ミトコンドリア遺伝子であるチトクロームcオキシダーゼ(CCO:cytochrome c oxidase)の欠損は加齢に伴って獲得される.組織化学染色によってCCOの欠損を検出することで固形臓器におけるクローン拡大が探索された.その結果,大腸,乳腺,皮膚などの臓器で正常組織においてもクローン拡大が観察され,大腸などいくつかの臓器では加齢とクローン拡大との関連が示された6).しかし,X染色体不活性化やCCOの欠損はクローン拡大を認識するためのマーカーにはなるが,これらそのものは細胞増殖に寄与しておらず,正常組織におけるクローン拡大の機序については長らく不明であった.
次世代シークエンサーの登場により網羅的ゲノム解析が可能となり,その後の技術の向上により少量の核酸から解析が可能となったことで,がんと比較して微小な正常組織における変異クローンを同定することが可能となった.正常細胞における変異の蓄積については長らく不明であったが,血液細胞における報告にはじまり7)8),オルガノイド培養技術を応用した単一細胞コロニーのシークエンス9)や,レーザーマイクロダイセクション10)や微小サンプリング11)による幹細胞-分化細胞単位でのシークエンスにより,さまざまな臓器において1細胞あたりの遺伝子変異負荷が決定された.これらの研究によって細胞あたりの遺伝子変異数は加齢とともに増加すること,また,アルコール・タバコ,紫外線,発がん物質への曝露といった環境因子によっても増加することが示され,たとえ正常細胞であっても時間経過や環境因子への曝露によってたえず遺伝子変異を蓄積し続けることが明らかとなった3).加齢や環境因子によって導入される遺伝子変異は,その原因毎に特徴的な塩基置換パターンを示すことが明らかになっており,変異シグネチャー解析(十時の稿)で解説いただいた.
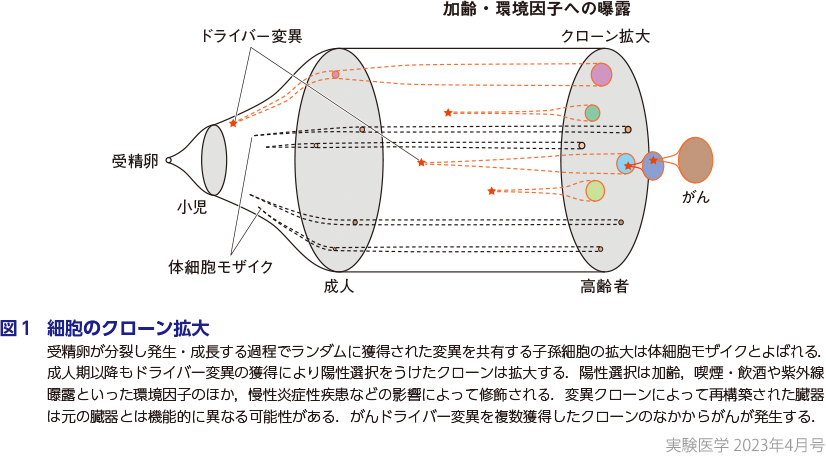
❷ 体細胞モザイクと陽性選択
個体発生の起源である1個の受精卵は細胞分裂をくり返して約40兆個にのぼる細胞からなる個体を形成する.また,個体のサイズが一定となる成人期以降もたえず細胞は入れ替わり続ける.その自然史のなかで,個々の細胞は変異を獲得すると同時に,多くの子孫細胞を発生させる場合もあれば,細胞死に至り消失する場合もある.ある時点で細胞が有した変異に着目し,その変異を共有する子孫細胞の集団をクローンとよぶ.個体を構成する細胞は受精卵を頂点し,変異によって分岐する枝であらわされた系統樹としてあらわすことができる.胎生期や小児期の間,細胞は変異をランダムに獲得しつつ総数を増やす.このような成長に伴うクローン拡大は体細胞モザイク(somatic mosaicism)とよばれるが(図1),ここでは変異は細胞増殖に対して中立である.成人期以降も細胞の入れ替わりによってクローンのサイズはランダムに変動しうるが,大幅なクローン拡大はドライバー変異の獲得によって達成される.ドライバー変異の概念はがんゲノム解析研究から生まれたが,正常組織においてもがんと同様に,細胞に増殖能もしくは細胞死への抵抗能を付与する変異として定義される.ドライバー遺伝子は細胞の種類や細胞が置かれた環境と密接に関係し,また,正常組織においてくり返し観察されることから,ドライバー変異クローンは陽性選択(positive selection)を受けると考えられる(図1).ドライバー遺伝子を同定する方法として,ランダムな変異率と観察された変異数の比較12)や,同義置換と非同義置換の期待値と観測値との差の比較を行うdN/dS解析13)などがある.
2正常組織におけるクローン進化
時に100年に及ぶヒトの生涯にわたって組織中の変異クローンを追跡することは現実的に不可能であり,また,観察のため一度採取してしまうと当該クローンが消失するという問題を克服するため,さまざまな年齢や環境因子を有する被験者を解析し比較することで正常組織におけるクローン進化が検討されてきた.代表的な臓器におけるクローン進化についての詳細は本誌の他稿に譲り,ここでは臓器間比較を行うことで得られる特徴を述べる.
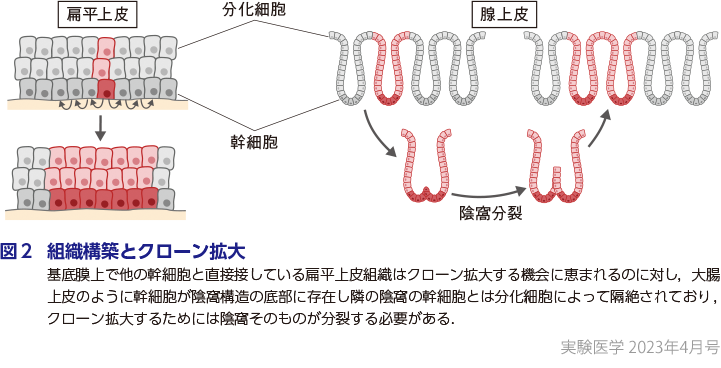
加齢とともにクローン拡大が観察される臓器として最初に報告されたのは造血器であり,若年者と比較して高齢者の末梢血において数%に及ぶ分画に拡大したクローンの存在が報告された(南谷の稿)7)8).これをクローン性造血とよぶが,拡大クローンの多くは急性骨髄性白血病や骨髄異形成症候群(MDS)などの血液悪性腫瘍で同定されるドライバー遺伝子に変異を有していた.このことから,造血器におけるクローン拡大は血液悪性腫瘍ドライバー変異の獲得によって駆動されることが判明した.この発見に続いて皮膚,食道(横山の稿),気管支(吉田の稿)などにおいて加齢に伴う,がんと共通するドライバー変異によって駆動されるクローン拡大が報告された.これとは対照的に,正常大腸では成長に伴う体細胞モザイクとしてのクローン拡大は観察されるが,ドライバー変異によるクローン拡大はほとんど存在しない.この違いは,組織中で幹細胞が他の幹細胞ニッシェに浮遊して移動しうる造血器や,基底膜上で他の幹細胞と直接接している扁平上皮組織はクローン拡大する機会に恵まれるのに対し,大腸上皮のように幹細胞が陰窩構造の底部に存在し隣の陰窩の幹細胞とは分化細胞によって隔絶されており,クローン拡大するためには陰窩そのものが分裂する必要があるという組織構築の違いに起因すると考えられる(図2).
古くから疫学研究によりがんのリスク因子として環境因子が明らかとなっている.環境因子に曝露されることで細胞の変異蓄積速度が加速するだけでなく,ドライバー変異の獲得とクローン拡大が促進されることが喫煙とクローン性造血,紫外線曝露と皮膚,飲酒・喫煙と食道上皮,喫煙と気管支といった組合わせで明らかとなった.このなかで特に興味深いのは,気管支上皮において,禁煙後の患者では喫煙患者と比較して,遺伝子変異負荷の大きいクローンが減少し,喫煙の影響が少なく遺伝子変異負荷が小さいクローンが再出現してクローンが交替し,さらにはTP53変異クローンの割合も減少することである14).このことは,50代までに禁煙すればその後の肺がん発症リスクが減少するという疫学的所見とも合致し,正常組織におけるがんドライバー変異クローンをコントールすることで発がん予防につなげられる可能性を示唆している.
クローン拡大を促進するその他の環境因子として慢性炎症があげられる.長期間,慢性炎症にさらされた臓器において,組織は破壊と再生をくり返すことで細胞の入れ替わりが促進される.大腸の慢性炎症性疾患である潰瘍性大腸炎(IBD)では,健常者の大腸とは大きく異なり,時に19 cmにわたって拡大する巨大なクローンが観察され,そのクローン進化の過程では炎症性シグナルへの応答を減弱させる変異が選択される(南木の稿)11)15).同様に,胃酸の逆流による食道の慢性炎症に起因するバレット食道でも広範な変異クローンの拡大が報告されている16)17).
慢性炎症とは異なるが破壊と再生をくり返す組織として子宮内膜があげられ,50歳になるとほぼすべての腺管がドライバー変異を獲得するに至ることが報告されている(山口らの稿)18).性ホルモンの影響は乳腺上皮でも観察され,詳細は西村の稿に詳しい.このようなクローン拡大の観察が可能になったのは,技術・理論の進歩によるところも大きい.本特集では,単一細胞由来検体を用いた解析(吉田の稿),SNPアレイデータを用いた遺伝的モザイシズムの解析(鎌谷・寺尾の稿)でそれぞれ解説いただいた.このように加齢やさまざまな環境因子の影響を受けて臓器ごとに特徴的なクローン拡大・陽性選択がなされるが,今後もさらに多くの臓器でその詳細が解明されると期待されている(図1).
3正常組織におけるクローン拡大の意義
正常組織で選択されるドライバー遺伝子の多くは対応する臓器のがんドライバー遺伝子と共通することから,正常組織における変異クローンはがんの起源と考えられている.従来から最初のがんドライバー変異が獲得される時期はがんと診断される数十年前に遡ると考えられてきたが19),食道の観察では,時に乳幼児期といった人生の早期にがんドライバー変異が獲得されることが明らかとなった(横山の稿)20).このことは,ドライバー変異そのものは環境因子に長期間暴露される前から組織中に生じうることを意味する.長期間にわたって環境因子と変異クローンとの相互作用によってクローン拡大し,そのなかから追加のドライバー変異を獲得したサブクローンが現れ,最終的にがんに至ると考えられる.正常組織とがんで観察されるドライバー変異の頻度を比較すると,正常組織の方が変異頻度が高い遺伝子変異(例えば潰瘍性大腸炎におけるNFKBIZ変異など)が存在し,このようなクローンそのものはがんへと進化し難い(=発がんのコンテキストでは陰性に選択される)ことが明らかとなり11),正常組織におけるクローン進化はがんのみに帰結しないことを示唆している(図3).また,正常組織とがんとの間で変異頻度が同程度の遺伝子は,従来のがんのゲノム解析ではがんドライバーと考えられて来たが,正常組織において選択を受けた遺伝子ががんに受け継がれた結果を見ているとも考えられ,発がんを積極的に促進しているわけではない可能性が考えられる3)(図3).NFKBIZ変異に代表される炎症関連遺伝子に変異によって再構築された大腸上皮は炎症性刺激への応答が変化しており11)15),このことは,加齢や環境因子に暴露された結果,変異クローンによって再構築された臓器は機能的に元の臓器とは異なると考えられ,加齢による臓器機能の低下や疾患の病態を修飾する因子となっている可能性がある.また,正常組織におけるクローン拡大が他臓器の疾患を惹起する例としてクローン性造血が動脈硬化・心血管イベントの原因となっていることが明らかになっており8)21)22),他臓器・疾患への関与についても今後のさらなる研究が期待される.

おわりに
正常組織におけるクローン拡大はその存在が明らかとなり,さまざまな臓器において報告されるに至っている.がんと共通する遺伝子変異があることから発がんの起源としての意義が唱えられているが,加齢に伴う臓器機能の低下や疾患への関与など,いまだ詳細が解明されていない点も多い.今後,技術発展によりクローン間の相互作用や,ゲノムおよびエピゲノム異常の協調進化などが明らかにされると期待される.本領域の発展ががんの予防や治療,老化や慢性疾患の克服に資することを願う.
文献
- Martincorena I & Campbell PJ:Science, 349:1483-1489, doi:10.1126/science.aab4082(2015)
- Bailey MH, et al:Cell, 173:371-385.e18, doi:10.1016/j.cell.2018.02.060(2018)
- Kakiuchi N & Ogawa S:Nat Rev Cancer, 21:239-256, doi:10.1038/s41568-021-00335-3(2021)
- Fialkow PJ:Biochim Biophys Acta, 458:283-321, doi:10.1016/0304-419x(76)90003-2(1976)
- Fey MF, et al:Blood, 83:931-938(1994)
- Taylor RW, et al:J Clin Invest, 112:1351-1360, doi:10.1172/JCI19435(2003)
- Genovese G, et al:N Engl J Med, 371:2477-2487, doi:10.1056/NEJMoa1409405(2014)
- Jaiswal S, et al:N Engl J Med, 371:2488-2498, doi:10.1056/NEJMoa1408617(2014)
- Blokzijl F, et al:Nature, 538:260-264, doi:10.1038/nature19768(2016)
- Lee-Six H, et al:Nature, 574:532-537, doi:10.1038/s41586-019-1672-7(2019)
- Kakiuchi N, et al:Nature, 577:260-265, doi:10.1038/s41586-019-1856-1(2020)
- Lawrence MS, et al:Nature, 499:214-218, doi:10.1038/nature12213(2013)
- Martincorena I, et al:Cell, 171:1029-1041.e21, doi:10.1016/j.cell.2017.09.042(2017)
- Yoshida K, et al:Nature, 578:266-272, doi:10.1038/s41586-020-1961-1(2020)
- Nanki K, et al:Nature, 577:254-259, doi:10.1038/s41586-019-1844-5(2020)
- Ross-Innes CS, et al:Nat Genet, 47:1038-1046, doi:10.1038/ng.3357(2015)
- Stachler MD, et al:Nat Genet, 47:1047-1055, doi:10.1038/ng.3343(2015)
- Moore L, et al:Nature, 580:640-646, doi:10.1038/s41586-020-2214-z(2020)
- Yachida S, et al:Nature, 467:1114-1117, doi:10.1038/nature09515(2010)
- Yokoyama A, et al:Nature, 565:312-317, doi:10.1038/s41586-018-0811-x(2019)
- Jaiswal S, et al:N Engl J Med, 377:111-121, doi:10.1056/NEJMoa1701719(2017)
- Fuster JJ, et al:Science, 355:842-847, doi:10.1126/science.aag1381(2017)
本記事のDOI:10.18958/7223-00001-0000394-00
著者プロフィール
垣内伸之:2007年,京都大学医学部医学科卒業.消化器内科医として臨床に従事する中でがん診療の限界を自覚し,’14年に京都大学大学院医学研究科(消化器内科学,腫瘍生物学)で研究を開始.がんの起源に迫る研究から個体内で細胞がさまざまな進化を遂げることを見出し,’19年より同助教を経て,’21年より独立し現職(京都大学白眉センター特定准教授)となる.さまざまな臓器のクローン進化をテーマとして研究を行っている.